The His-Tag: Fundamentals And Principles
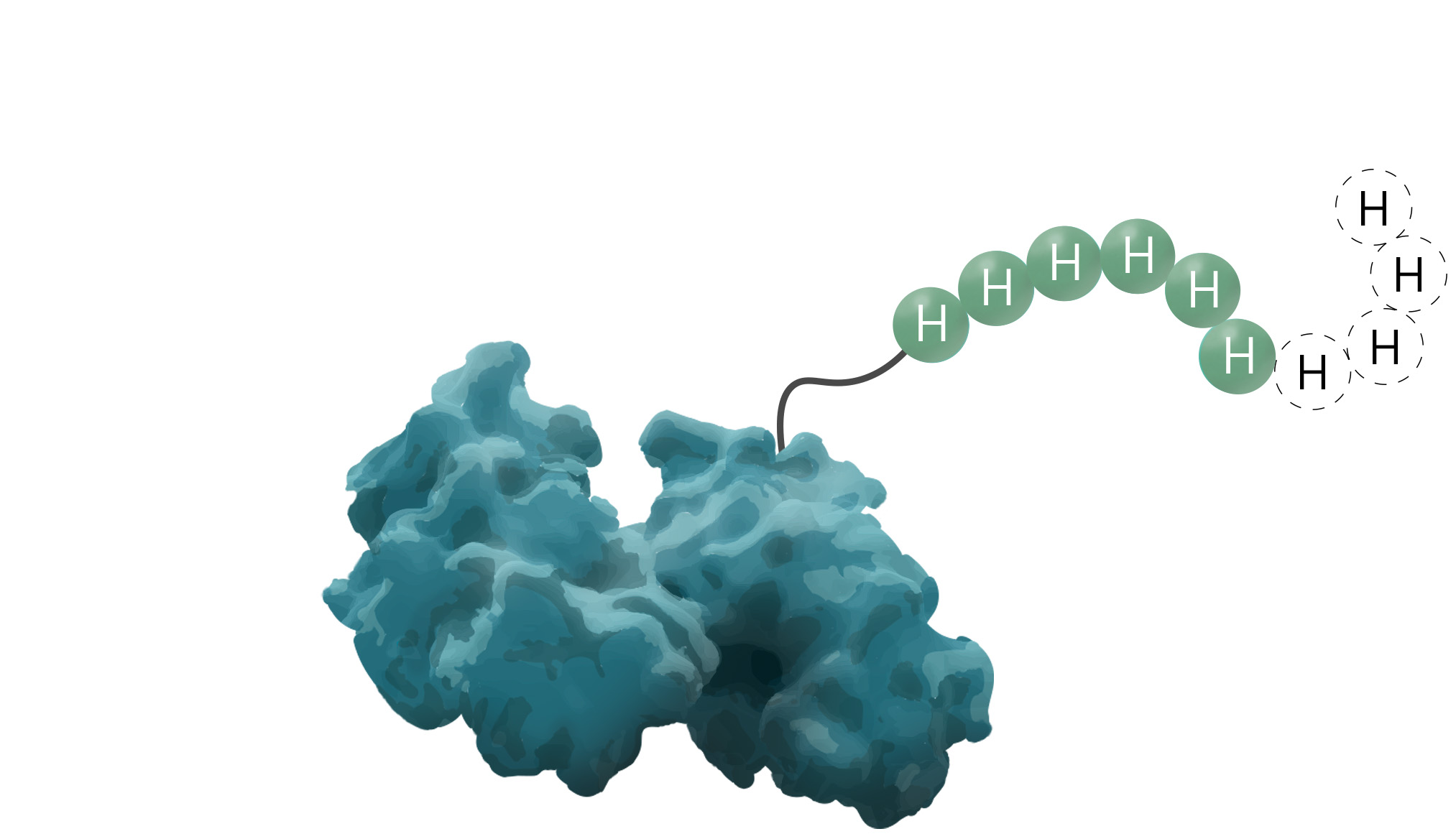
The polyhistidine- or His6-tag is a protein tag originally developed for efficient protein purification in 1988 (Hochuli, Bannwarth & Döbeli et al., 1988). As a result of its comparably great cost-benefit ratio, it is known as one of the most widely used protein affinity tags to date. Due to its small size, variable between four to ten amino acids, it seldom interferes with the target protein's function. Additionally, its low immunogenicity and versatility under native and denaturing conditions are elevating this protein tag to stand out among others. The underlying purification principle of the His-tag is an interaction between metal ions and the imidazole ring of histidine.
The Features
Table 1: Overview of the most important aspects of the protein affinity tag
| Features of the His-tag: | |||
|---|---|---|---|
| Amino Acid Sequence | HHHHHH (4-10) | ||
| DNA Sequence | 5'-CAT CAC CAT CAC CAT CAC-3' | ||
| Size | 840.8 Da (His6) | ||
| Compatibility to recombinant proteins | Can be added either to the N- or C-Terminus of a protein | ||
| Specificity of interaction (KD) | 10 µM | ||
| Protein yield per ml of high-quality purification resin | Up to 80 mg / ml (GFP-His) | ||
| Elution conditions | Imidazole, histidine, or pH shift (pH 4-5) | ||
How does a His-tag work?
The purification of a protein via his-tagging is achieved through an interaction between varying divalent metal ions, like Ni2+ (the predominantly used ion) and the basic imidazole ring of histidine. His-tags are able to bind to those metals at a neutral to slightly basic pH (7.5 – 8 is typical). It is worth noting that the binding affinity between those two interaction partners is proportional to the length of the His-tag. A tenfold higher binding affinity is achieved with His10-tags compared to His6-tags (Guignet, Hovius & Vogel, 2004; Fessenden, 2009). Nevertheless, as a first trial, it is advised to minimize the number of histidine residues in order to avoid possible perturbations of protein function (Bornhorst & Falke, 2000).
During the later stages of the purification process, the protein can be eluted by decreasing the pH to 4-5, although it should be noted that some proteins denature at such a low pH. Furthermore, the majority of scientists prefer an elution by competition with higher concentrations of imidazole (100-500 mM). It is also advised to insert low molarities of imidazole (5-20 mM, rarely up to 80 mM) into the loading and washing buffers to reduce nonspecific binding events, as some endogenous proteins or natural occurring histidines are able to display weak binding to IMAC resins (Bolanos-Garcia & Davies, 2006). Optimal conditions for every protein should always be determined individually.
During the later stages of the purification process, the protein can be eluted by decreasing the pH to 4-5, although it should be noted that some proteins denature at such a low pH. Furthermore, the majority of scientists prefer an elution by competition with higher concentrations of imidazole (100-500 mM). It is also advised to insert low molarities of imidazole (5-20 mM, rarely up to 80 mM) into the loading and washing buffers to reduce nonspecific binding events, as some endogenous proteins or natural occurring histidines are able to display weak binding to IMAC resins (Bolanos-Garcia & Davies, 2006). Optimal conditions for every protein should always be determined individually.
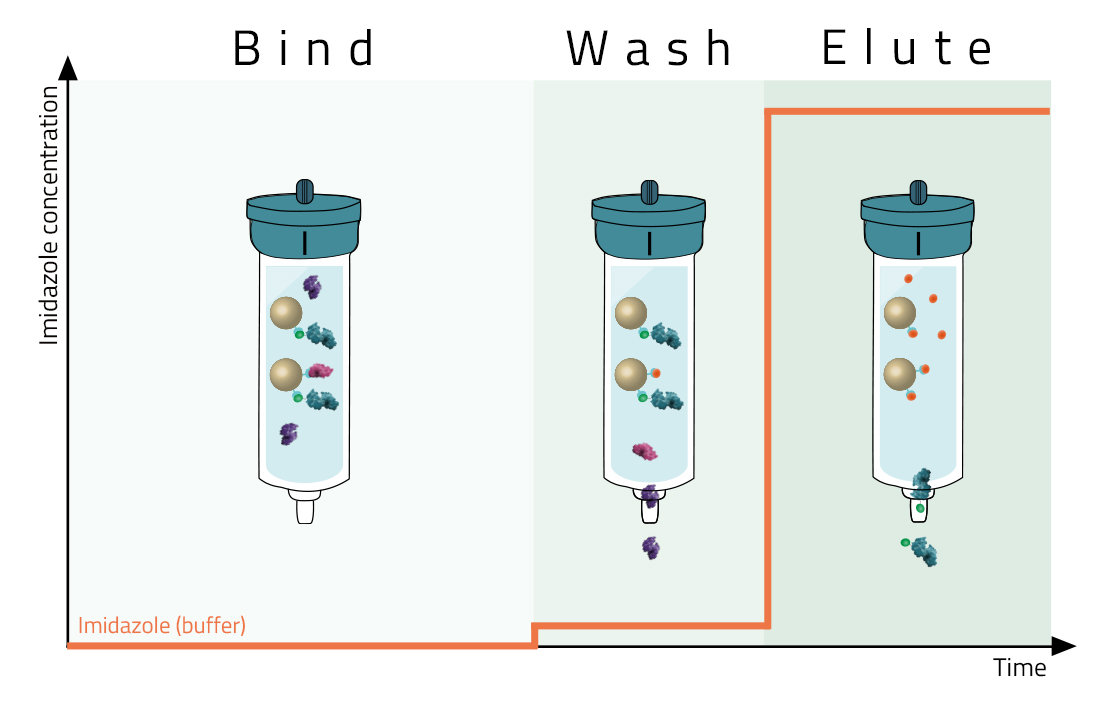
Most affinity purification protocols follow the same three steps (Fig. 2):
1. Binding:
A crude solution containing the His-tagged protein is applied to the column and binds based on the affinity tag - matrix interaction.
2. Washing:
Other proteins which bind unspecifically are washed away with suitable washing buffers (see magenta protein in Fig. 2). These buffers should already contain low molarities of imidazole.
3. Elution:
Specifically bound protein is eluted from the column, typically by competitive binding of a similar molecule (e.g. histidine and imidazole), by cutting off the tag with a protease, or by destabilization of the affinity tag - matrix interaction e.g. by a change of pH. The pure eluate can then be collected in reaction tubes for further research.
Protein Purification via His-tag
How do you purify a protein with a His-tag?
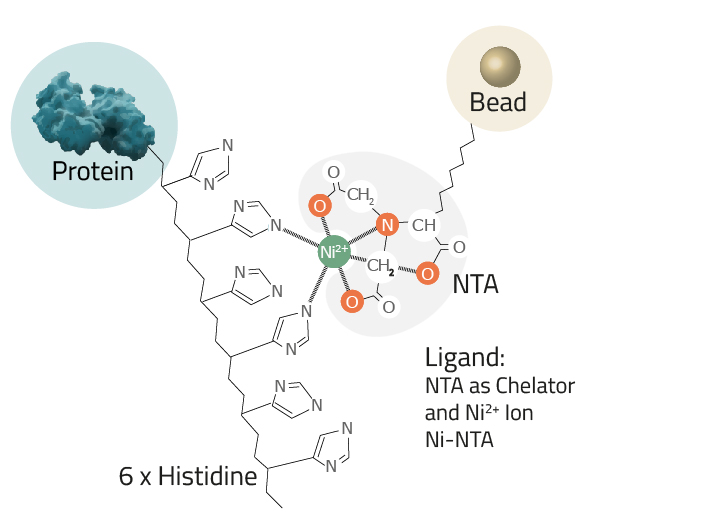
Protein purification via His-tag is often reached by an immobilized metal affinity chromatography or in short IMAC. This is a widely-used purification method to separate proteins and peptides that show an affinity for metal ions (like Ni2+ or Co2+) from a solution. His-tagging a protein enables the newly formed fusion proteins to bind with the chelated metals, resulting in substantial enrichments in very few steps. Common chelators utilized as ligands are IDA and NTA-agarose resins. They can easily be paired with a divalent metal, depending on the desired affinity/specificity ratio of the process (see Fig. 4).
Table 2: Comparison between three ligands and their nickel binding capacities
| Ligand | Coordination sites | Nickel binding |
|---|---|---|
| Iminodiacetic acid (IDA) | 3 | Weak |
| Nitrilotriacetic acid (NTA) | 4 | Moderate |
| INDIGO-Ni | 5 | Strong |
Choosing the correct type of His-tag purification resin or magnetic bead is based on three factors
- The Ligand: This is the molecule to which the metal ion is coupled in a chelator complex (see Fig. 3).
Cube Biotech offers three different ligands:- NTA: Nitrilotriacetic acid is the most commonly used ligand. Its chelator complex with nickel is depicted in Figure 3.
- IDA: Iminodiacetic acid is the second most commonly used one. Learn the differences between IDA and NTA here.
- INDIGO: This ligand was developed here at Cube Biotech. Among its advantages are a highly increased DTT and EDTA tolerance. Find out more about this novel ligand.
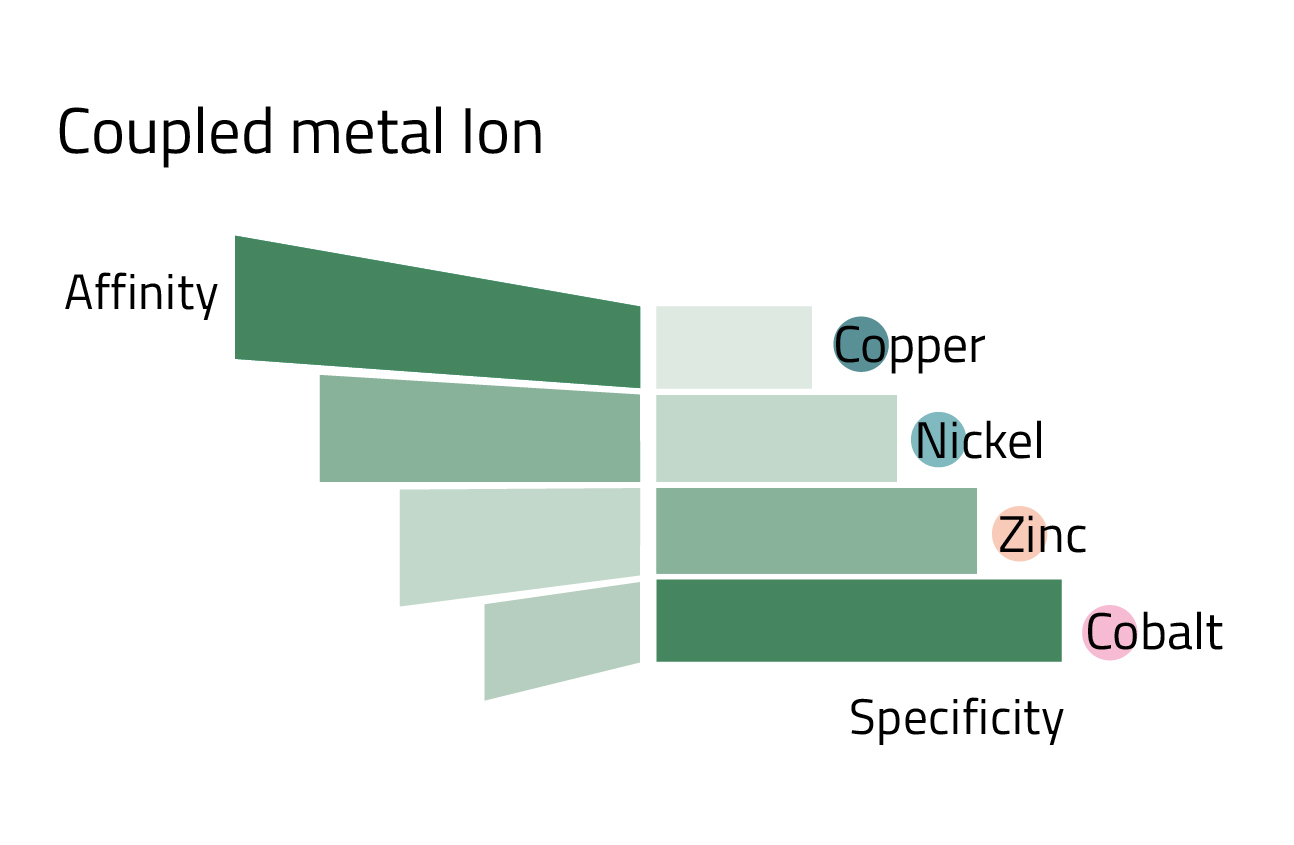
- The coupled metal ion: Depending on which metal ion is coupled to the ligand, the affinity, and specificity of the His-tag purification changes. Copper has the highest affinity, while in turn having the lowest specificity. Cobalt marks the other end of the spectrum. Nickel, the most commonly used metal ion has a good balance between affinity and specificity. Rule of thumb: The higher the protein yield, the less specific the purification becomes. Figure 4 can give you an overview of the affinity/specificity ratio of the most commonly used metal ions for His-tag purification.
- The size of the beads: The bigger the used beads are in diameter the higher the flow rate for FPLC purification. This renders them more useful for viscous cell media or batch purification. However, making use of a slower flow rate usually increases the protein yield, as smaller beads have a higher surface-to-volume ratio. Therefore, the same volume of resin can bind more protein than beads of bigger sizes. We offer bead diameters of 30 µm, 40µm, 100 µm, and XL sized (~400 µm). Further sizes are available on request.
Chelator complex structure and metal ions compared:
How to add a His-tag
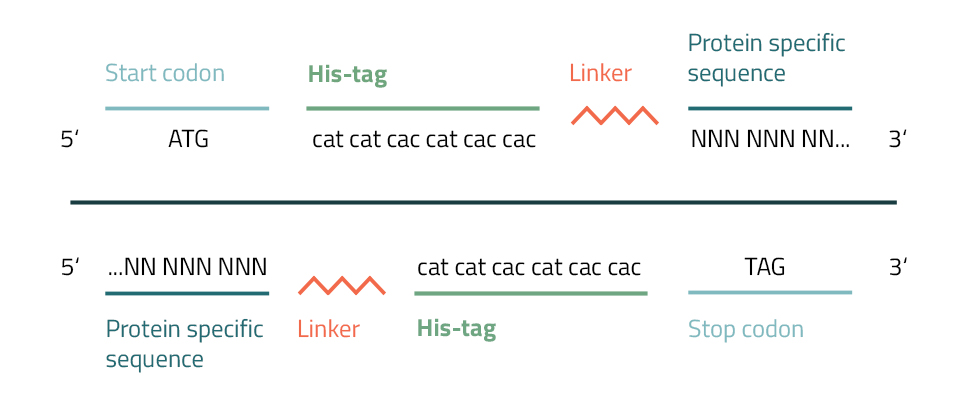
The addition of a Polyhistidine affinity tag to a protein can be accomplished by various methods. Their small size and charge enable them to be easily integrated into target genes by PCR or site-directed mutagenesis.
To accomplish this you will need to create a primer with repetitive histidine codons (CAC/CAT) next to a START or STOP codon. Furthermore, several bases from one end of your protein of interest are added to this fragment. The newly incorporated hexahistidine affinity tag will approximately add 0,9 kDa to your molecular weight. To prevent interference with the protein activity caused by the tagging, linkers are inserted between tag and protein (see Fig. 5). They are divided into three general groups: flexible linkers, rigid linkers, and in vivo cleavable linkers (Chen, Zaro & Shen, 2013). Common linkers of the flexible group are gly-gly-gly or gly-ser-gly. It is advised, to vary their lengths or to leave them out completely in order to achieve the best results for your specific protein.
To accomplish this you will need to create a primer with repetitive histidine codons (CAC/CAT) next to a START or STOP codon. Furthermore, several bases from one end of your protein of interest are added to this fragment. The newly incorporated hexahistidine affinity tag will approximately add 0,9 kDa to your molecular weight. To prevent interference with the protein activity caused by the tagging, linkers are inserted between tag and protein (see Fig. 5). They are divided into three general groups: flexible linkers, rigid linkers, and in vivo cleavable linkers (Chen, Zaro & Shen, 2013). Common linkers of the flexible group are gly-gly-gly or gly-ser-gly. It is advised, to vary their lengths or to leave them out completely in order to achieve the best results for your specific protein.
It is likewise possible to clone code fragments of His-tags, created from synthetic oligonucleotides, into a chosen location of a plasmid (David, Gee & Andersen et al., 1997). Commercially, there is a broad variety of cloning vectors available on the market for the incorporation and expression of His-tagged proteins in varying expression systems (e.g vectors for E. coli) (Skerra, 1994). They enable you to directly subclone target DNAs into vectors that are containing polyhistidine sequences along with adjacent protease cleavage sites for easy disposal of the affinity tag after purification (see Fig. 6). Please note, that when cloning, it is important to stay in the correct reading frame in order to achieve the best results.
1. Vector containing N-terminal polyhistidine sequence (orange) / multiple cloning site (light blue) / vector backbone (green)
2. Addition of a protein-specific sequence (PSS) flanked by restriction sites
3. Cloning of your code fragment into the fusion vector
2. Addition of a protein-specific sequence (PSS) flanked by restriction sites
3. Cloning of your code fragment into the fusion vector

His-tag Applications
Besides their well-known use as protein purification aids, His-tags play a role in many further applications of biotechnology. Notable examples are pulldown assays, assistance in crystal formation, or methods of immune labeling.
- Pulldown assays: affinity tags like His6 or GST paired with an immobilized ligand (e.g. Ni2+ agarose resin) can be used to specifically bind to a target protein complex in order to purify and identify it (Louche, Salcedo & Bigot, 2017)
- Crystal formation: an abundance of proteins can be crystallized with an intact His-tag without a negative impact on the tagged target protein (Carson et al., 2007). Smits et al. even described cases where the His-tag assisted in the formation of crystals in 2008 (Smits, Mueller & Grieshaber, 2008)
- Immune labeling: hexahistidine tags are utilized in ELISA assays, Western blot detection, flow cytometry, immunohistochemistry, or immune fluorescence
- Serial His-tagging: single subunits of a large protein can be tagged respectively to act as a marker for activity states in a gel (Howarth, Chinnapen & Gerrow et al., 2006)
- SDS-PAGE staining: metal-based fluorescence dyes facilitate the detection of a His-tagged peptide in a gel (Zhao, Hellman & Zhan et al., 2010)
- SPR immobilization strategies: surface plasmon resonance, used to determine kinetics and thermodynamics of two binding molecules, offers multiple possible immobilization strategies for protein attachment via His-Tag (Fischer, Leech, & Hubbard, 2011)
Frequently asked Questions
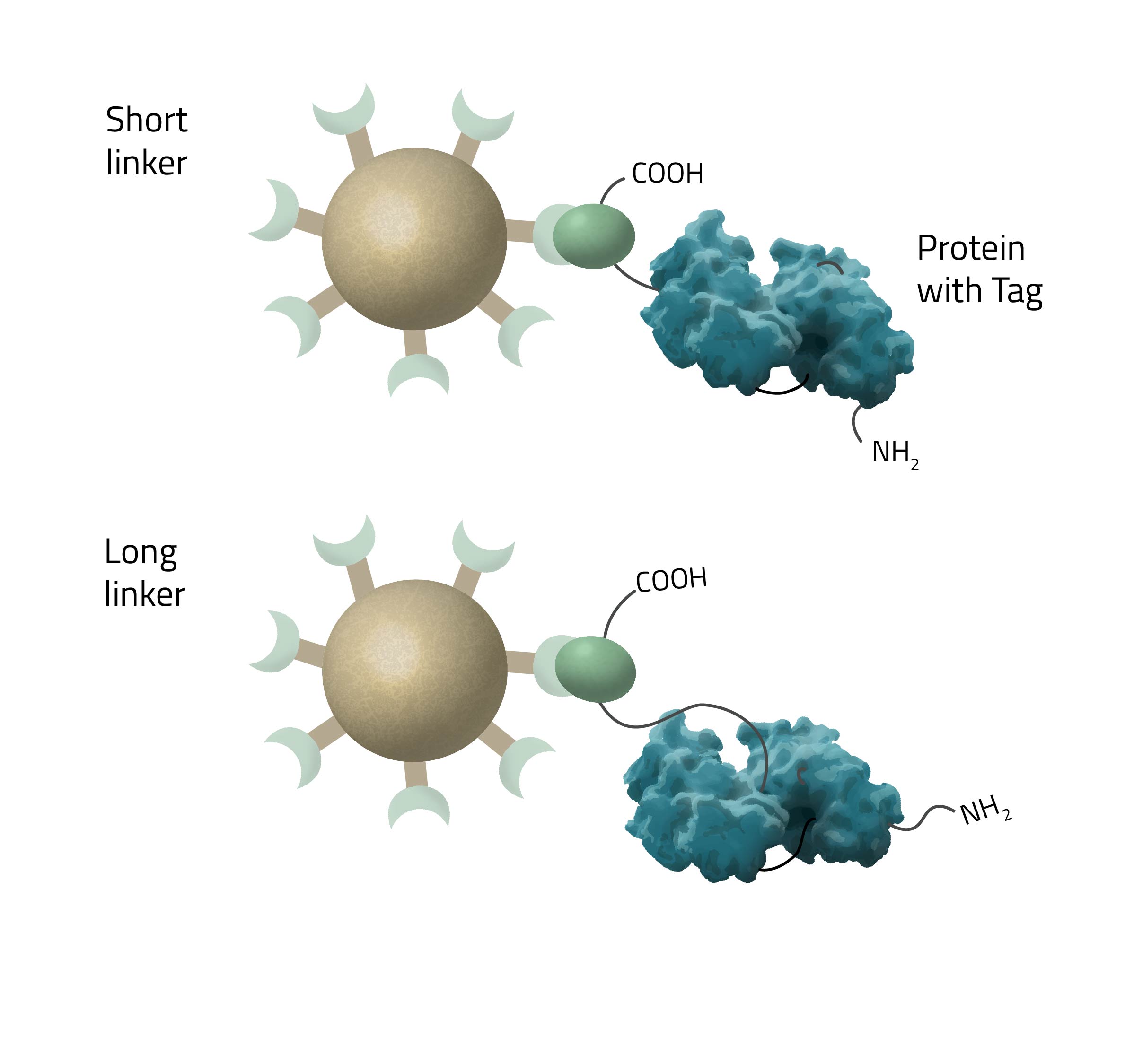
How long should a linker to the His-tag be?
The length of the linker/spacer depends on the structural accessibility of the target protein. Longer chain lengths have higher mobility and are thus crucial for sterically hindered tags (see Fig. 7).
Where should I place the His-tag?
The His-tag can be added either to the N- or C-terminus. Optimal placement is protein specific and dependent on molecular structure as some sites might perturb the accessibility of the tag.
How to cleave off a His-tag?
The affinity tag can be removed by use of a protease cleavage site inserted between polyhistidine tag and protein (Nikolov, Hu & Lin et al., 1992).
Why cleave the His-tag?
The His-tag is sometimes cleaved off to ensure unimpaired protein activity and easier crystallization, but no general rule can be applied here. Due to the relatively small size and charge His-tags seldom interfere with protein activity. Furthermore, there are even described cases of His-tag-assisted crystal formation. However, for pharmaceutical applications and usage, proteins are often required to be in their native state, which is why cleaving of the His-tag is a standard procedure there.
How to elute His-tags from beads?
Elution from the beads can be achieved with the help of increased imidazole concentrations, histidine, or with a pH shift (pH 4 - 5).
References
- Hochuli, E., Bannwarth, W., Döbeli, H. et al. Genetic Approach to Facilitate Purification of Recombinant Proteins with a Novel Metal Chelate Adsorbent. Nat Biotechnol 6, 1321–1325 (1988).
- Guignet, E.G., Hovius, R., Vogel, H. Reversible site-selective labeling of membrane proteins in live cells. Nature Biotechnology, 22 (4), pp. 440-444 (2004)
- Fessenden, JD., Förster resonance energy transfer measurements of ryanodine receptor type 1 structure using a novel site-specific labeling method. PLoS One, 4 (10), p. e7338 (2009)
- Bolanos-Garcia, V.M., Davies, O.R. Structural analysis and classification of native proteins from E. coli commonly co-purified by immobilised metal affinity chromatography. Biochim. Biophys. Acta, 1760, pp. 1304-1313 (2006)
- David, N.E., Gee, M., Andersen, B. et al. J Biol, Chem 272:15553 (1997).
- Skerra A. A general vector, pASK84, for cloning, bacterial production, and single-step purification of antibody Fab fragments. Gene, 141(1), 79–84 (1994).
- Petty, K J., Metal-chelate affinity chromatography. Current protocols in neuroscience vol. Chapter 5 (2001)
- Carson, M. et al., His-tag impact on structure. Acta crystallographica. Section D, Biological crystallography vol. 63, Pt 3 (2007)
- Smits, S.H., Mueller, A., Grieshaber, M.K., Schmitt, L. Coenzyme- and His-tag-induced crystallization of octopine dehydrogenase Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun., 64, pp. 836-839 (2008)
- Howarth, M., Chinnapen, DF., Gerrow, K. et al. A monovalent streptavidin with a single femtomolar biotin binding site. Nat Methods 3, 267–273 (2006)
- Chen, X., Zaro, J.L., and Shen, W.-C. Fusion protein linkers: property, design and functionality Adv. Drug Delivery Rev. 65, 1357– 1369 (2013)
- Bornhorst, J. A. & Falke, J. J. Purification of proteins using polyhistidine affinity tags. Methods in enzymology, 326, 245–254 (2000)
- Louche A., Salcedo S.P., Bigot S. Protein–Protein Interactions: Pull-Down Assays. Bacterial Protein Secretion Systems. Methods in Molecular Biology, vol 1615 (2017)
- Zhao, C., Hellman, L.M., Zhan, X., Bowman, W.S., Whiteheart, S.W., Fried, M.G. Hexahistidine-tag-specific optical probes for analyses of proteins and their interactions. Analytical Biochemistry, Vol. 399, Issue 2 (2010)
- Fischer, M., Leech, A. P. & Hubbard, R. E. Comparative assessment of different histidine-tags for immobilization of protein onto surface plasmon resonance sensorchips. Analytical chemistry, 83(5), 1800–1807 (2011)
- Nikolov, D. B., Hu, S. H., Lin, J. et al. Crystal structure of TFIID TATA-box binding protein. Nature 360, 6399 (1992)
